In this study, BCPDA was used for the solid-phase TRF IA study. It overcomes the disadvantages of the need to enhance the solution, the environmental helium ion contamination, and the ability to measure only in the liquid phase. This simplifies the measurement procedure. Combined with BCPDA labeled BSA, the protein content in the labeling process was determined. Provide theoretical basis and experimental techniques for the TRF IA system. Materials and Method
1 Materials
1. 1 Instrument spectrophotometer, nucleic acid protein detector, magnetic stirrer, pH22 acidity meter, chromatography column, Kjeldahl nitrogen determination instrument, semi-automatic nitrogen determination instrument.
2. 2 Reagents BCPDA (Self-synthetic), 99. 999%Eu2 03 (homemade), Bovine Serum Albumin, Sephadex G250 Dextran, Tris-hydroxymethylaminomethane (Super Pure), Coomassie Brilliant Blue G2250, Anhydrous Carbonic Acid The reagents such as sodium and anhydrous sodium bicarbonate are all commercially available and analytically pure. The relevant buffer solutions are all homemade. Experimental water: Deionized water.
2 methods
2.1 Labeling reaction After the protein is derivatized in a carbonate buffer of 0.1 mol/L pH = 9.1 at room temperature, the newly prepared BCPDA is dissolved in dimethylformamide (DMF) solution and divided into several portions. Add 1min intervals, while swirling the protein solution continuously, stirring at room temperature for 30min.
2.2 Separation, purification, and labeling In addition to the binding of BCPDA to proteins, free BCP2DA is also present in the reaction solution. The labeled reaction solution is passed through a gel column and the bound BCP2DA is separated from the free BCPDA. For the binding peak, the second peak is the free BCPDA peak. The portion of the binding peak was collected under monitoring by a Nucleoprotein Analyzer 280nm.
2. 3 Preparation of BCPDA standard solution Weigh a certain amount of BCP2DA dissolved in DMF, dilute to a certain concentration with a pH = 7.8 Tris2HCl buffer solution, and prepare a BCPDA standard solution.
2.4 Determination of BCPDA content in solution 1 The characteristic absorption at 325 nm using BCPDA has an extinction coefficient of 1.5 × 104 mol/L-1 ·m-1. The concentration of BCPDA is calculated as follows: BCPDA concentration (C) = Absorbance (A) / (cell thickness × extinction coefficient) × dilution factor, in this experiment: C = A / (1cm × 1. 5 × 104mol/L-1m-1) × dilution factor; 2 Separate the BCPDA standard solution Diluted to different concentrations with pH = 9. 1 carbonate buffer, pH = 8.0 NH4HCO3 buffer, pH = 7.8 Tris2HCl buffer, and set it to a 1cm × 1cm quartz cell and measured its A value at 325nm. Make a working curve. The sample was measured in the same manner and the BCPDA concentration in the sample was found on the corresponding working curve.
2. 5 BSA standard solution preparation Weigh a certain amount of BSA dissolved with ethanol, dilute to a certain concentration with carbonate buffer solution of pH = 9.1, set aside.
2. 6 Coomassie brilliant blue dye preparation Weigh 0. 020g coomassie brilliant blue G2250 dissolved in 95% 10mL ethanol, plus 85% phosphoric acid 20mL, diluted with deionized water to 200mL.
2. 7 Coomassie brilliant blue staining BSA standard solution to prepare different volumes of BSA standard protein solution, were added to a 10mL volumetric flask, with Tris2HCl buffer added to 1mL, add the staining solution to volume, shake immediately, at room temperature for 10min , absorbance at 595 nm.
2. 8 Coomassie brilliant blue staining BSA-BCPDA solution prepared from the reaction flask to take a certain amount of reaction solution to a 5mL volumetric flask, add Tris2HCl buffer to 1mL, then add Coomassie brilliant blue G2250 staining solution to the scale, room temperature for 10 minutes, Measure absorbance at 595 nm.
Results and discussion
Absorption spectra of 1 BSA
The BSA stock solution was diluted to different concentrations with pH = 9. 1 carbonate buffer, pH = 8. 0NH4HCO3 buffer, pH = 7.8 Tris2HCl buffer, respectively, and set at 1cm × 1cm quartz cell at 280nm. Absorbance value. The three buffers showed a good linear relationship between BSA absorbance and its concentration (1028 to 1026 mol/L).
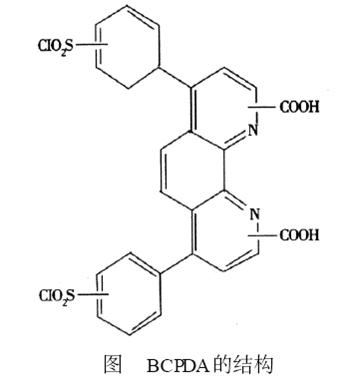
2 BCPDA Structure
From the structural point of view of BCPDA, it contains two sulfonyl chloride groups, which can be covalently bound to the free amino groups of the protein under mild conditions. The heteroaromatic nitrogen it contains and the two carboxyl groups together form the site of chelation with the phosphonium ion. The BCPDA2Eu3 + chelate labeled protein has formed a very stable fluorescence chelate, which can produce higher quantum yield, larger Stokes shift, narrower emission band and longer fluorescence lifetime (10~ 1000 μs) of fluorescence. Its structure is shown in the figure.
Absorption spectra of 3 BCPDA
The BCPDA standard solution was diluted to different concentrations with pH = 9. 1 carbonate buffer, pH = 8. 0 NH 4HCO 3 buffer, pH = 7.8 Tris 2 HCl buffer, and set in a 1cm × 1cm quartz cell and measured at 280°C. The 325 nm A value showed a good linear relationship between BCPDA diluted with the three buffers and its concentration (1026-1024 mol/L). The main absorption peaks were 232.95, 291.58, and 325.04 nm.
4 Absorption of BSA at 280 nm in BCPDA
The determination of protein content in the TRFIA labeling experiment involves the calculation of the labeling ratio, the monitoring of the labeling reaction process, and the control of the labeling conditions. Therefore, the determination of the protein content and its accuracy and sensitivity are particularly important. The dual-function chelating agent such as EDTA in the liquid phase TRF IA system does not interfere with the characteristic absorption of proteins at 280 nm, and the protein content can be determined by the UV absorbance method at 280 nm. The solid-phase system established on the basis of the new dual-function chelating agent BCPDA, BCPDA and protein have strong absorption at 280 nm, because the characteristic absorption of the protein at 280 nm is due to the large number of amino acids and benzene ring structure generated by hydrolysis. However, from the molecular structural formula of BCPDA, there is also a benzene ring structure and contains a carboxyl group, so the determination of protein content by ultraviolet absorption method is not applicable to the solid phase system.
5 Coomassie brilliant blue G2250 staining assay protein content contrast experiment
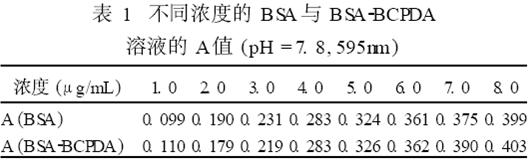
The same concentration of BSA standard solution and BSA2BCPDA labeled reaction solution were measured by Coomassie brilliant blue G2250 staining method. The comparison results are shown in Table 1. The results showed that BCPDA did not interfere with the determination of BSA in the range of 1.0 to 8.0 μg/mL.
The experiments were also conducted within a concentration range of 5.0-40 μg/mL. The A values ​​of BSA and BSA2BCPDA solutions had a good agreement, which accorded with Beer's law and met the requirements of marker monitoring.
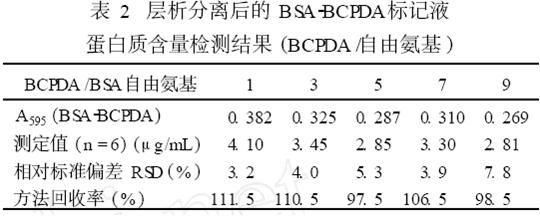
6 Protein content of BSA2BCPDA labeling solution after chromatographic analysis was detected by Coomassie brilliant blue method from the collected purified reaction solution, and was compared with the standard curve of BSA standard solution to find out the corresponding protein content and calculate the total amount recovered. The BSA content of BSA2BCPDA in different samples (BCPDA/free amino) was determined experimentally, and the results are shown in Table 2.
7 buffer solution selection
Experiments using pH = 7.8 Tris2HCl, pH = 9. 1NH4HCO3, pH = 9.1 carbonate, pH = 8.0 NH4HCO3 buffer as a solvent system were determined using a pH = 7.8 Tris2HCl buffer solution. The linearity of the work curve is better than the latter three.
8 related sample parameters
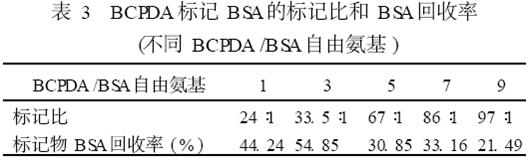
The labeling ratios (BSA mol/BCPDA mol) and BSA recovery (%) of the different (BCPDA/BSA free amino) loading ratios were determined experimentally using a 1.5 cm×25 cm column (see Table 3). The highest protein recovery of BCPDA-labeled BSA reaction was 54.85%, and the presumed marker ratio increased with the increase of BCPDA.
Chelation and fluorescence spectra of 9 BCPDA2BSA2 Eu3 +
A certain concentration of BCPDA2BSA solution was added Tris2HCl buffer containing Eu3 +, diluted a certain number of times, and incubated at 37 ° C for 0. 522h, BCPDA and Eu3 + chelate reaction, stable at room temperature 1h with 337.1 nm excitation, Measured fluorescence peaks of erbium ion 589, 611 nm.
in conclusion
(1) UV absorption method is not suitable for the determination of protein in the solid phase TRFIA system BSA2BCPDA.
(2) No significant absorption was observed at 595 nm when a suitable amount of Coomassie brilliant blue staining solution was added to the 1027 to 1025 mol/L BCPDA standard solution.
(3) Determination of BSA in BSA2BCPDA by Coomassie brilliant blue G2250 staining method. Under the experimental conditions, the working curve ranged from 0.50 to 40. 0 μg/mL, and the curve regression equation was: Y =0.00032 +0. 0015X , r=0.996. The lower limit of detection was calculated as 3σ/r, which was 0.35 μg/mL (n = 9) and RSD < 7.8%. The method is simple and rapid, has the advantages of less sample consumption, less interference, and high sensitivity. (4) The Coomassie brilliant blue staining method was used for protein determination in solid phase TRF IA markers. It has certain significance for the study of polyphenylene chelates, ligands and protein linkages.